Genomics
Metagenomics
Metagenomics projects aim to caracterize the content of a microbial community found in a given environment. These communities can be found in multiple environments such as human gut, soil, plant root system, oceanic water, etc. Often, these communities are caracterized by determining the bacterial species present in one or more sample as well as their abundance. Multiple DNA sequencing technologies can be used to extract DNA present in a sample and computational methods for caracterizing the communities are often specialized to the sequencing technology of DNA extraction method used (16S rRNA or Whole Genome Shotgun).
Design of a machine learning approach for bacterial microorganisms detection in metagenomic sequencing without alignment procedures
Master's thesis research project in DialloLab at UQAM
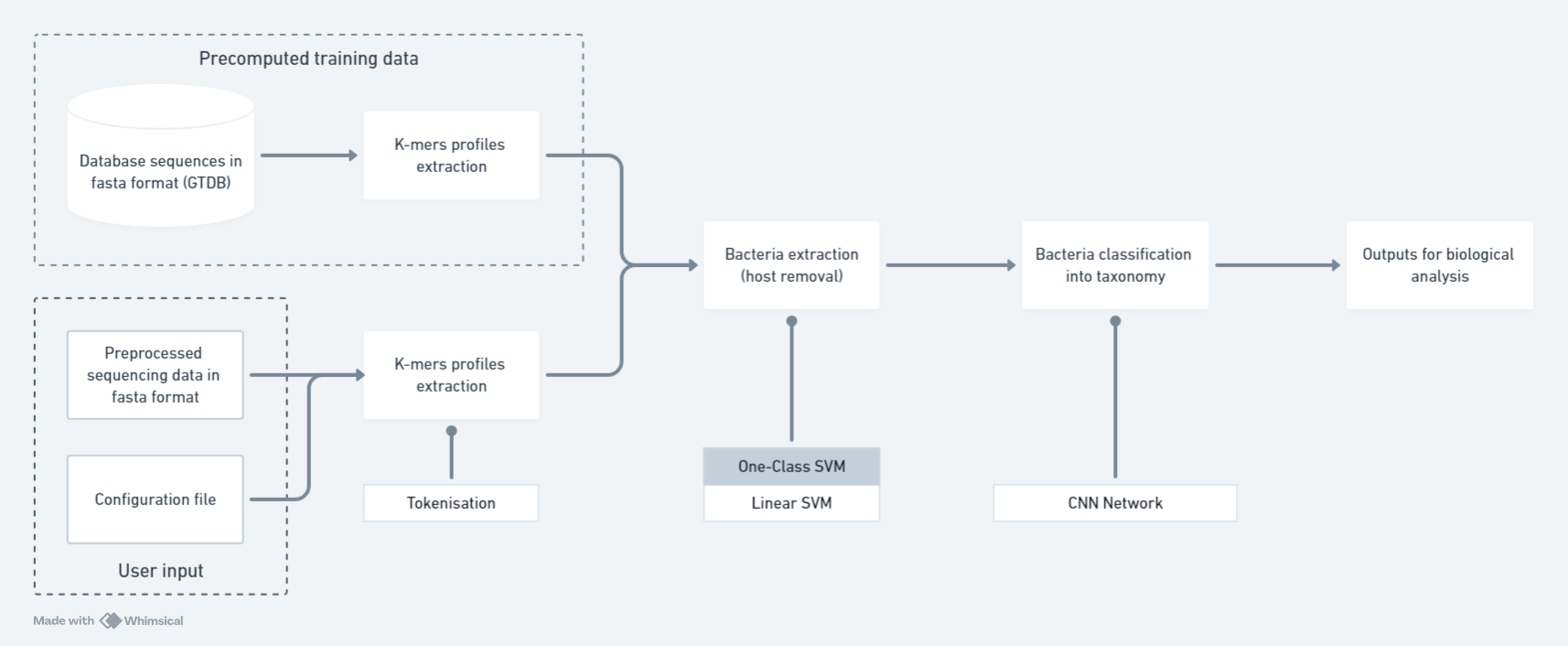
Design of a machine learning approach for bacterial microorganisms detection in metagenomic sequencing without alignment procedures - Master's thesis research project in DialloLab at UQAM
Project description:
Data from metagenomic sequencing experiments are complex to analyse because of their size, knowledge contained in databases and performances of already available tools, based on multiple criteria. The most precise methods are are based on alignment procedures, but usually require large computationnal resources. Therefore, this research project aims to design and implement a method for classifying metagenomic sequencing data at a large scale using alignment-free methods. The research work is conceptual in nature but it was also implemented to test it's performances on artificial data. The main part of the project is concentrated on large scale data handling and machine learning. To permit training models on whole genomes and subsequent alignment-free classification of metagenomic sequencing data, the k-mers method for profiles extractions was used. Classification is done in a reccurent manner using a "top-down" strategy. Therefore, a binary classification permits to extract the bacterial sequences then the multiclass classification is done on multiple taxonomic levels for the sequences that were identified at a previous level, going from the largest level to the most precise one. For each step two classical algorithms and three neural networks are trained on whole genomes from the "Genome Taxonomy Database" and tested using cross validation on reads simulated from training genomes.
Tools & packages used:
- Python
- Tensorflow
- Keras
- Scikit-Learn
- Ray
- Pandas
- Numpy
- PyArrow
- Biopython
- InSilicoSeq
Responsibilities:
- Design
- Implementation
- Testing
Other contributors to the project:
- Amine M. Remita
- Steven W. Kembel (co-director)
- Abdoulaye Baniré Diallo (co-director)
 Code repo
Code repo
Metagenome analysis of data from a scientific article
DESS degree project
Metagenome analysis of data from a scientific article - DESS degree project
Project description:
This is a session project for two classes at UQAM. An article was chosen to fetch data from and was then analyzed using an alternate pipeline to familiarize with CLI genomic analysis tools and recreate results obtained in the base article.
- Analyse the microbiota using the Mothur analysis pipeline and tools on data found in the article for the course BIF-7104
- Make a taxonomic relations tree using the identified species in a subjects microbiota which would be use for further biological analysis for the course BIF-7101
Tools & packages used:
- Mothur pipeline
- MetaWrap pipeline
- Narval cluster (Digital Research Alliance of Canada)
- Kraken
- R
- Bash
Responsibilities:
- Retrieve the raw sequencing data
- Quality control of the data
- Curate the data according to QC
- Run the Mothur analysis pipeline on the data
- Run the MetaWrap analysis pipeline on the data
- Use HPC an server from the Digital Research Alliance of Canada for the analysis
- Assemble genomes present in the sequencing data
- Plot the results of the analysis
Other contributors to the project:
- Abdellatif El Ghizi
 Article
Article
Transcriptomics
Transcriptomics aim to analyse the expression of genes in a given sample taken from one or more individuals. These analysis can be run on RNA or DNA depending on the research interest and the sequencing technology used. For each sequencing types, certain methods exist for processing and analyzing data.
WGS analysis pipeline development
DESS degree internship in DialloLab at UQAM
WGS analysis pipeline development - DESS degree internship in DialloLab at UQAM
Project description:
Project in collaboration with Silversides Veterinary research laboratory for the CERMO-FC. Identify genetic variants causing birth malformations causing prematured death in Biewer Terrier dog breed. The data available were DNA-seq sequencing of prematurely dead individuals and their parents.
Tools & packages used:
- Bash
- Python
- FastP
- BWA
- Sambamba
- Picard
- GATK
- BCFTools
- Mutect2
- VarScan
- ANNOVAR
Responsibilities:
- Development of the data analysis pipeline
- Execution of the genetic comparison analysis pipeline
Other contributors to the project:
- Golrokh Kiani
Monocyte Gene and Molecular Expression Profiles Suggest Distinct Effector and Regulatory Functions in Beninese HIV Highly Exposed Seronegative Female Commercial Sex Workers
Contract with Roger Lab at CR-CHUM & UdeM
Monocyte Gene and Molecular Expression Profiles Suggest Distinct Effector and Regulatory Functions in Beninese HIV Highly Exposed Seronegative Female Commercial Sex Workers - Contract with Roger Lab at CR-CHUM & UdeM
Project description:
We have previously reported that the female genital tract (FGT) of Beninese HIV highly-exposed seronegative (HESN) commercial sex workers (CSWs), presented elevated frequencies of a myeloid HLA-DR+CD14+CD11c+ population presenting “tolerogenic” monocyte derived dendritic cells (MoDC) features. In order to assess whether a differential profile of monocytes may be involved in the generation of these genital MoDCs, we have herein characterized the blood monocyte compartment of Beninese HESNs (HIV-uninfected ≥ 10 years CSWs) and relevant controls (HIV-uninfected 2.5–5 years CSWs herein termed “early HESNs”), HIV-infected CSWs, and low-risk HIV-uninfected women from the general population. Transcriptomic analyses by RNA-Seq of total sorted blood monocytes demonstrate that in comparison to the control groups, HESNs present increased expression levels of FCGR2C, FCAR, ITGAX, ITGAM, CR2, CD68, and CD163 genes, associated with effector functions. Moreover, we found increased expression levels of genes associated with protection/control against SHIV/HIV such as CCL3, CCL4, CCL5, BHLHE40, and TNFSF13, as well as with immune regulation such as IL-10, Ahr, CD83, and the orphan nuclear receptor (NR)4A1, NR4A2, and NR4A3. Through multicolor flow cytometry analyses, we noticed that the frequencies of intermediate and non-classical monocyte populations tended to be elevated in the blood of HESNs, and exhibited increased expression levels of effector CD16, CD11c, CD11b, as well as regulatory HLA-G, IL-10, and IFN-α markers when compared to HIV-uninfected women and/or HIV-infected CSWs. This profile is compatible with that previously reported in the FGT of HESNs, and likely confers an enormous advantage in their resistance to HIV infection.
Tools & packages used:
- R
- tidyverse
- ggplot2
- gtable
- ggpubr
- digest
Responsibilities:
- RNA-seq data analysis between four human populations
- Statistical analysis of flow cytometry data produced by the team
- Graphics and layouts. Everything was done in R.
Other contributors to the project:
- Laurence Blondin-Ladrie
- Lyvia Fourcade
- Alessandro Modica
- Matheus Aranguren
- Johanne Poudrier
- Michel Roger
A First Insight into North American Plant Pathogenic Fungi Armillaria sinapina Transcriptome
Bachelor's degree internship in Desgagné-Pénix Lab at UQTR
A First Insight into North American Plant Pathogenic Fungi Armillaria sinapina Transcriptome - Bachelor's degree internship in Desgagné-Pénix Lab at UQTR
Project description:
Armillaria sinapina, a fungal pathogen of primary timber species of North American forests, causes white root rot disease that ultimately kills the trees. A more detailed understanding of the molecular mechanisms underlying this illness will support future developments on disease resistance and management, as well as in the decomposition of cellulosic material for further use. In this study, RNA-Seq technology was used to compare the transcriptome profiles of A. sinapina fungal culture grown in yeast malt broth medium supplemented or not with betulin, a natural compound of the terpenoid group found in abundance in white birch bark. This was done to identify enzyme transcripts involved in the metabolism (redox reaction) of betulin into betulinic acid, a potent anticancer drug. De novo assembly and characterization of A. sinapina transcriptome was performed using Illumina technology. A total of 170,592,464 reads were generated, then 273,561 transcripts were characterized. Approximately, 53% of transcripts could be identified using public databases with several metabolic pathways represented. A total of 11 transcripts involved in terpenoid biosynthesis were identified. In addition, 25 gene transcripts that could play a significant role in lignin degradation were uncovered, as well as several redox enzymes of the cytochromes P450 family. To our knowledge, this research is the first transcriptomic study carried out on A. sinapina.
Tools & packages used:
- R
- ggplot2
- tidyr
- plotly graphs
Responsibilities:
- RNA-seq data analysis between two culture mediums
- Data vizualisation and exploration
- Graphics and layouts
Other contributors to the project:
- Narimene Fradj
- Natacha Mérindol
- Fatima Awwad
- Yacine Boumghar
- Hugo Germain
- Isabel Desgagné-Penix
RNA-Seq de Novo Assembly and Differential Transcriptome Analysis of Chaga (Inonotus obliquus) Cultured with Different Betulin Sources and the Regulation of Genes Involved in Terpenoid Biosynthesis
Bachelor's degree internship in Desgagné-Pénix Lab at UQTR
RNA-Seq de Novo Assembly and Differential Transcriptome Analysis of Chaga (Inonotus obliquus) Cultured with Different Betulin Sources and the Regulation of Genes Involved in Terpenoid Biosynthesis - Bachelor's degree internship in Desgagné-Pénix Lab at UQTR
Project description:
Chaga (Inonotus obliquus) is a medicinal fungus used in traditional medicine of Native American and North Eurasian cultures. Several studies have demonstrated the medicinal properties of chaga’s bioactive molecules. For example, several terpenoids (e.g., betulin, betulinic acid and inotodiol) isolated from I. obliquus cells have proven effectiveness in treating different types of tumor cells. However, the molecular mechanisms and regulation underlying the biosynthesis of chaga terpenoids remain unknown. In this study, we report on the optimization of growing conditions for cultured I. obliquus in presence of different betulin sources (e.g., betulin or white birch bark). It was found that better results were obtained for a liquid culture pH 6.2 at 28 °C. In addition, a de novo assembly and characterization of I. obliquus transcriptome in these growth conditions using Illumina technology was performed. A total of 219,288,500 clean reads were generated, allowing for the identification of 20,072 transcripts of I. obliquus including transcripts involved in terpenoid biosynthesis. The differential expression of these genes was confirmed by quantitative-PCR. This study provides new insights on the molecular mechanisms and regulation of I. obliquus terpenoid production. It also contributes useful molecular resources for gene prediction or the development of biotechnologies for the alternative production of terpenoids.
Tools & packages used:
- R
- ggplot2
- tidyr
- plotly graphs
Responsibilities:
- RNA-seq data analysis between three culture mediums
- Data vizualisation and exploration
- Graphics and layouts
Other contributors to the project:
- Narimene Fradj
- Karen Cristine Gonçalves dos Santos
- Fatima Awwad
- Yacine Boumghar
- Hugo Germain
- Isabel Desgagné-Penix